We have designed the Phonon workflow to automate the computation and
analysis of phonons of a periodic system.
This type of project starts with the definition of the atomic structure. We
refer the user to section Structure Modeling in ASAP for further information on ASAP structure
builder/viewer interface.
After the atomic structure is defined, edit the type of project by selecting
Phonons from the list of possible project types implemented in ASAP.
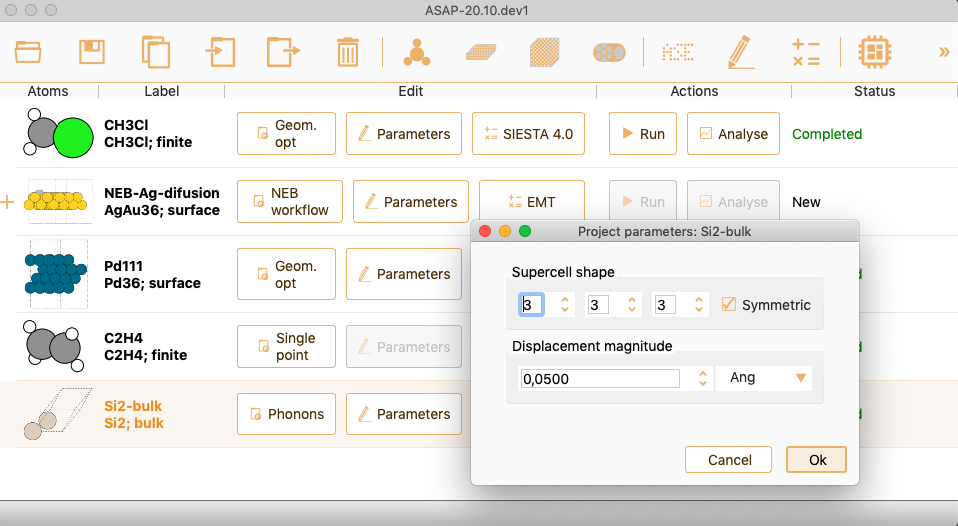
Then click on the Parameters icon to open the Phonon parameters widget.
You can tune the following project parameters:
Supercell shape. Supercell size used to calculate the force constant
matrix. Check Symmetric box to build a symmetric supercell.
Displacement magnitude. The distance the atoms are displaced to
calculate the force constant matrix. Select units between Ang, nm or Borh.
Click on the Calculator icon to select the code.
We refer the user to chapter Calculators for further information on ASAP available
calculators.
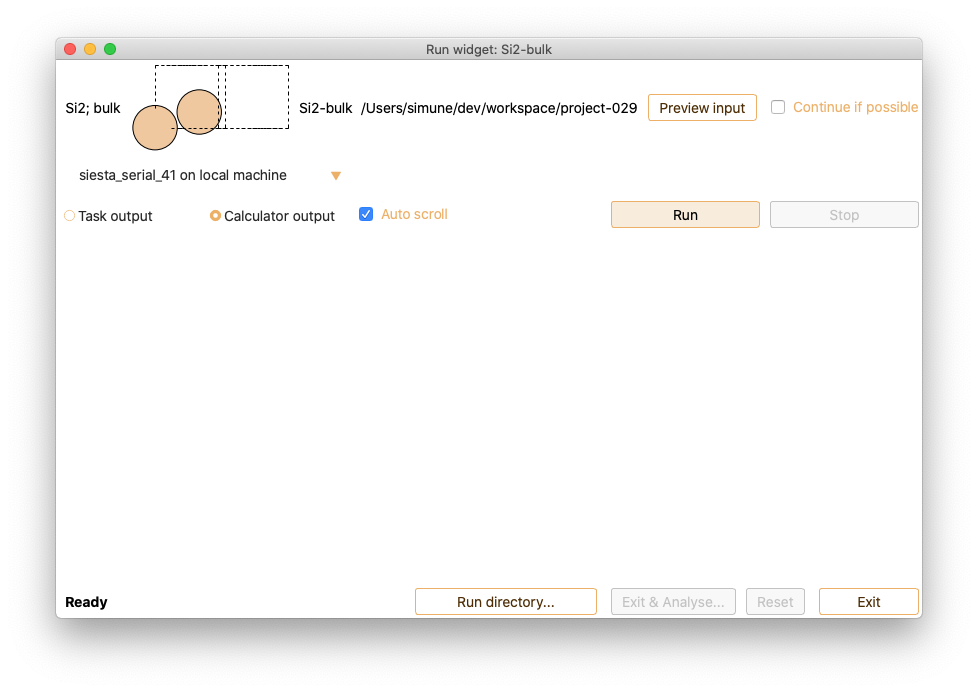
Click on the Run button to execute the calculation. Please be aware that
phonon calculations with SIESTA may take a significant amount of time to
complete.
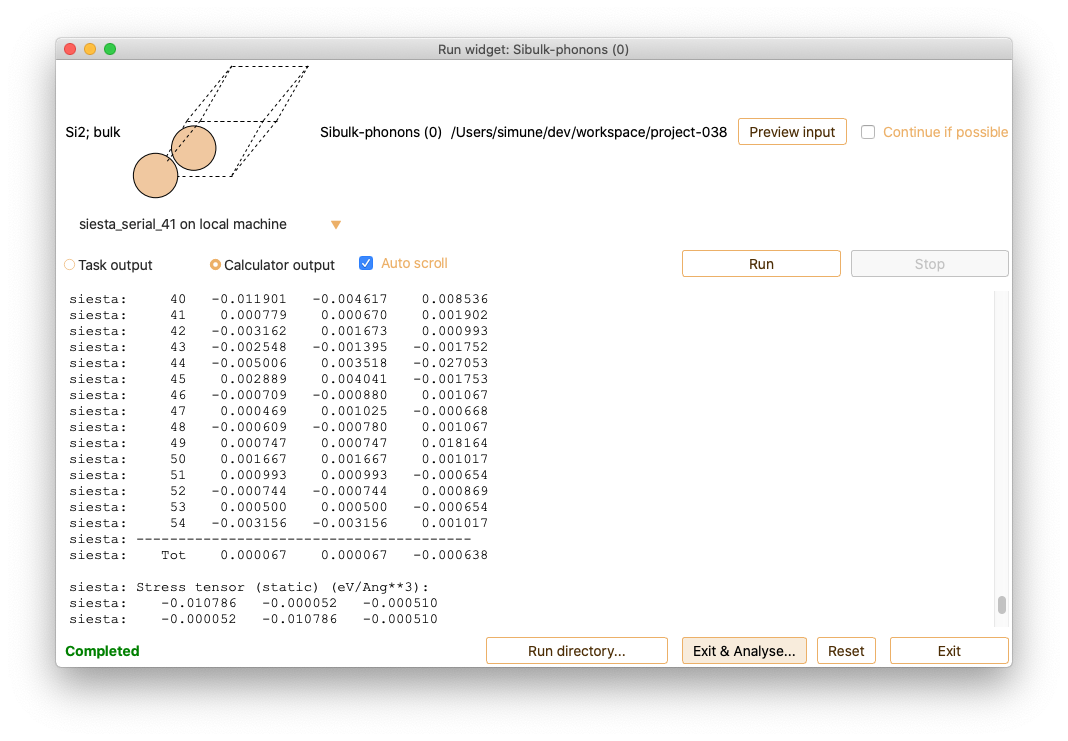
After the Phonon calculation is completed, click on the Exit and analyse
button to open the analysis widget.
It is also possible to open the analysis widget by selecting the Analyse
icon associated with a completed project,
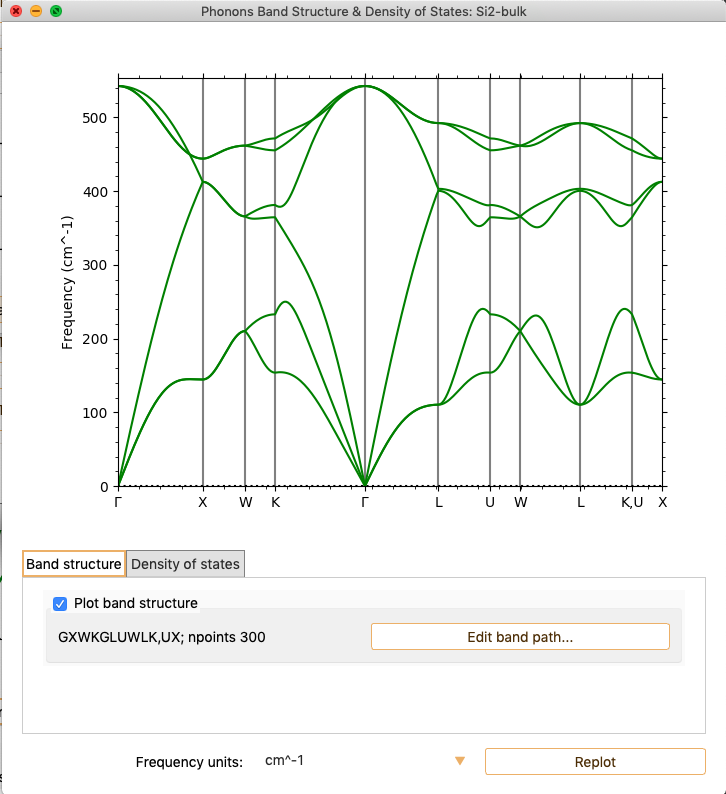
By default, the analysis widget only shows the Phonon Band Structure.
The energy unit (Y-axis) used by default is cm\(^{-1}\). You can
change it to meV or THz with the parameter Frequency units.
If you want to add new points to the band path, click on the button Edit
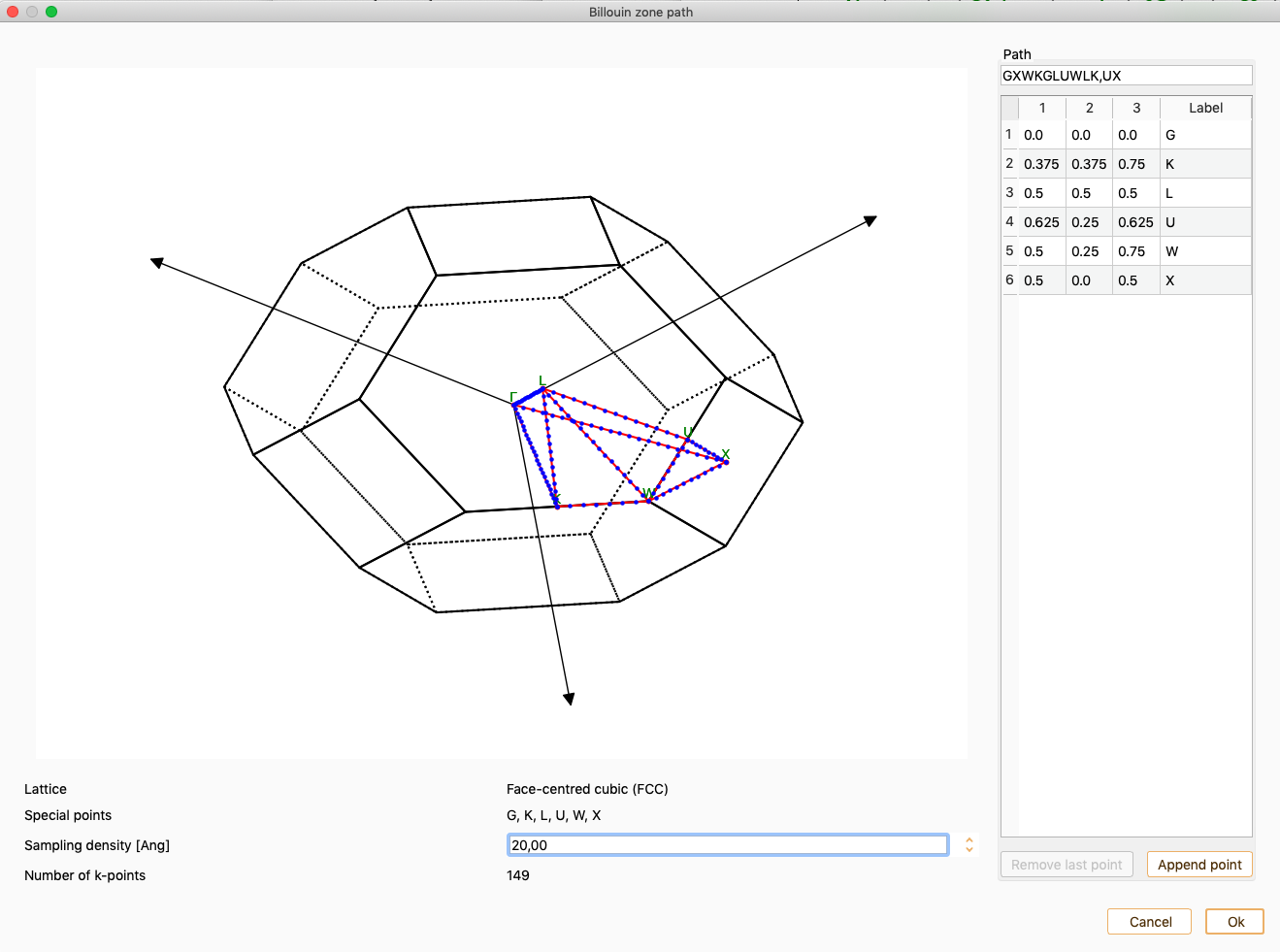
band path…. The widget shows the unit cell Brillouin zone, the band path
and the high symmetry k-points.
ASAP recognises the lattice type for a few Bravais lattice types. In this
case, the high symmetry k-points are labelled automatically. The automatic
k-point labelling currently supports the following Bravais lattice types:
Primitive cubic
Face-centred cubic (fcc)
Body-centred cubic (bcc)
Primitive tetragonal
Body centred tetragonal
Primitive orthorhombic
The widget also provides the following information:
Lattice. Bravais lattice type.
Special points. The high symmetry k-points.
Number of k-points. The total number of k-points that will be used
along the whole selected k-path.
You can edit the parameter Sampling density to modify the density of the
k-points.
Click the button Append point to add a new k-point to the band path.
Then, edit the k-point coordinates and label and add the new label to the
path.
Press the button OK and click on the button Replot to update the
figure.
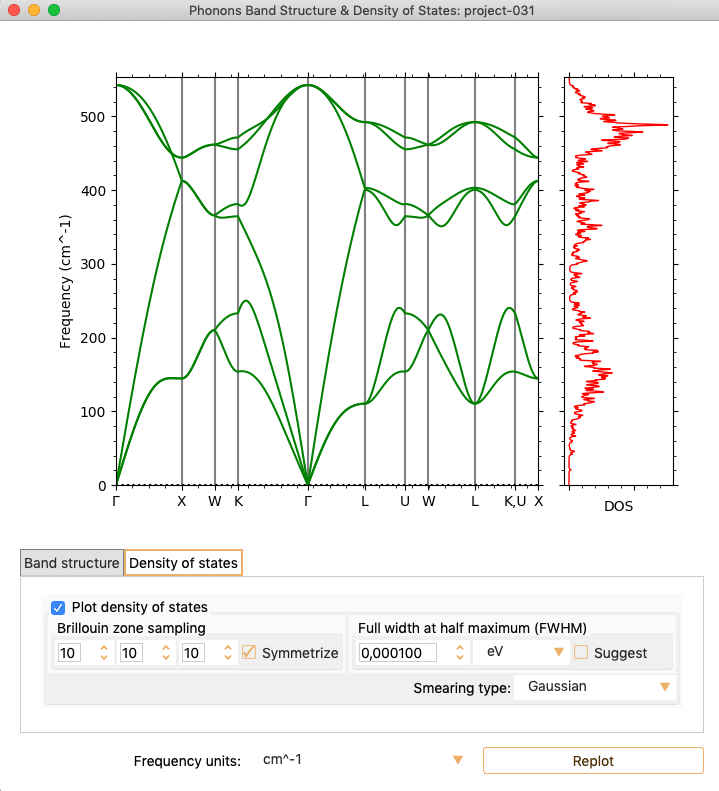
To visualise Density Of States in the same figure, click in the tab
Density of states, check Plot density of states box and click on the
button Replot.
Notice that the energy unit (Y-axis) is cm\(^{-1}\). You can change it
to meV or THz with the parameter Frequency units.
You can also tune the following parameters:
Brillouin zone sampling: Number of k-points used to plot the DOS.
Full width at half maximum (FWHM): The distance used between the
calculated points to perform the smearing.