Introduction#
Ready to use packages with necessary libraries and solvers.
Interactive GUI widgets for system construction, visualisation, and analysis.
Cross Platform performance: Linux, Mac, Windows operating systems.
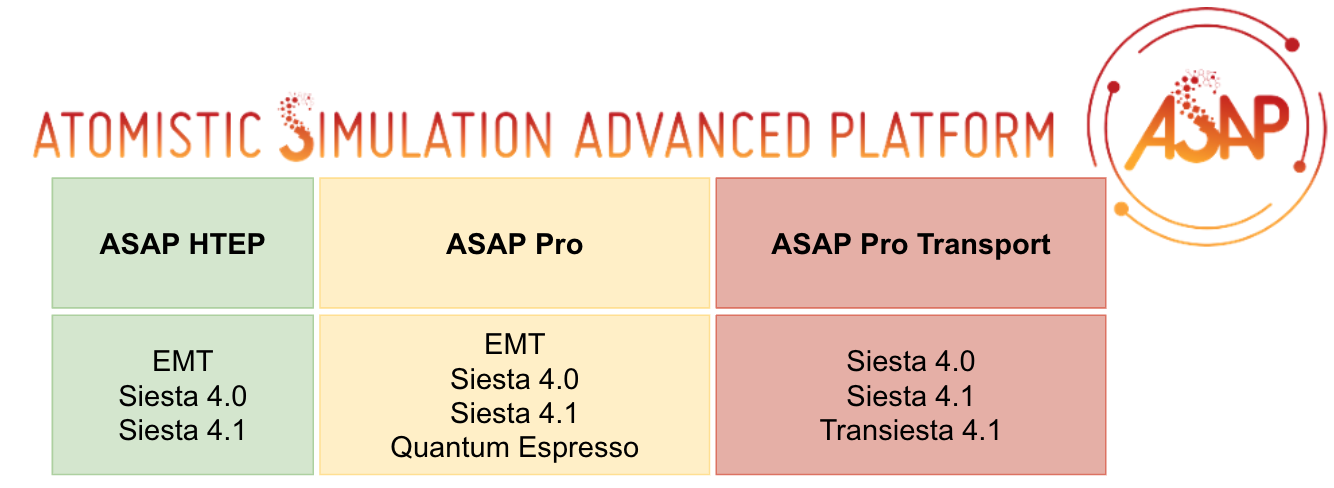
ASAP Subproducts#
Structure Builder Features |
ASAP HTEP |
ASAP Pro |
ASAP Pro Transport |
|---|---|---|---|
Import/View: pre-existing structures, structure manipulation, measure geometric quantities, dynamic visualisation, flexible view settings |
X |
X |
X |
Build (1D, 2D, 3D): molecular structures from built-in databases, most common crystal structures, supercell slabs (Miller index), nanoparticles, nanoribbons, natubes |
X |
X |
X |
Merge two structures |
X |
X |
X |
Build (electronic device): electrodes, buffer and scattering regions |
X |
Project Type: |
ASAP HTEP |
ASAP Pro |
ASAP Pro Transport |
|---|---|---|---|
X |
X |
X |
|
X |
X |
X |
|
X |
X |
X |
|
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
X |
||
X |
|||
X |
|||
X |
ASAP HTEP |
ASAP Pro |
ASAP Pro Transport |
|
|---|---|---|---|
Electronic properties: |
|||
Fermi energy |
X |
X |
X |
Single particle energies |
X |
X |
X |
Density Of States (DOS) |
X |
X |
X |
Partial Density of States (PDOS) |
X |
X |
X |
Band structure |
X |
X |
X |
Projected molecular orbitals (LDOS) |
X |
X |
X |
Charge (Mulliken, Hirshfeld, Voronoy, Bader) |
X |
X |
|
Electrostatic potential (incl. visualisation) |
X |
X |
|
Thermodynamics properties: |
|||
Equilibrium volume |
X |
X |
|
Bulk modulus |
X |
X |
|
Chemical properties: |
|||
Interaction energy |
X |
X |
|
Reaction diagram |
X |
X |
|
Activation energies |
X |
X |
|
Phonons and vibrations: |
|||
Vibrational spectrum |
X |
X |
|
Zero-Point Energy correction (ZPE) |
X |
X |
|
Phonon density of states |
X |
X |
|
Phonon band structure |
X |
X |
|
Chemical properties: |
|||
Interaction energy |
X |
X |
|
Reaction diagram |
X |
X |
|
Activation energies |
X |
X |
|
MD analysis: |
|||
Kinetic and Potential energy |
X |
X |
|
Radial Distribution Function (RDF) |
X |
X |
|
MDS, RSMD analysis |
X |
X |
|
Diffusion coefficients |
X |
X |
|
Velocity autocorrelation function |
X |
X |
|
Electronic nanotransport: |
|||
Transmission function |
X |
||
I-V curve |
X |
||
Conductance |
X |
About ASAP#
We have developed ASAP in Python 3. It is compatible with Python \(\ge\) 3.6. We use the Python library PySide2 (binding of the GUI toolkit Qt) for GUI rendering. The other main packages and libraries ASAP relies on are:
The Atomic Simulation Environment, aka ASE (https://wiki.fysik.dtu.dk/ase)
SciPy (https://scipy.org/)
Matplotlib (https://matplotlib.org/)
E. Artacho, D. Sánchez‐Portal, P. Ordejón, A. Garcia, and J. M. Soler, Linear‐Scaling ab‐initio Calculations for Large and Complex Systems, Phys. Status Solidi B 215, 809 (1999).
J. M. Soler, E. Artacho, J. D. Gale, A. Garcia, J. Junquera, P. Ordejón and D. Sánchez-Portal, The SIESTA method for ab initio order-N materials simulation J. Phys. Condens. Matter 14, 11, (2002)
E. Artacho, E. Anglada, O. Diéguez, J. D. Gale, A. Garcia, J. Junquera, R. M. Martin, P. Ordejón, J. M. Pruneda, D. Sánchez-Portal and J. M. Soler, The SIESTA method; developments and applicability, J. Phys. Condens. Matter 20, 064208, (2008).
A. H. Larsen, J. J. Mortensen, J. Blomqvist, I. E. Castelli, R. Christensen, M. Dulak, J. Friis, M. N. Groves, B.Hammer, C. Hargus, E. D. Hermes, P. C. Jennings, P. B. Jensen,K. Kaasbjerg, J. Kermode, J. R. Kitchin, E. L. Kolsbjerg, J. Kubal, S. Lysgaard, J. B. Maronsson, T. Maxson, T. Olsen, L. Pastewka, A. Peterson, C. Rostgaard, J. Schiøtz, O. Schutt, M. Strange, K.Thygesen, T. Vegge, L. Vilhelmsen, M. Walter, Z. Zeng, and K. W.Jacobsen, The Atomic Simulation Environment — A Python library for working with atoms, J. Phys. Condens. Matter 29, 273002 (2017).